recount3: human and mouse public RNA-seq data
1 Data
recount3 provides processed RNA-seq data for human and mouse in file formats similar to recount2 (Figure 1.1), which at its core is based on coverage bigWig files and exon-exon junction counts (see Raw file for more details). These two raw files power the whole recount3 ecosystem. In recount3, we have provided coverage count files for several human and mouse annotations with samples grouped by study. Some large studies, like GTEx and TCGA have been fragmented at the tissue level to make the data more accessible.
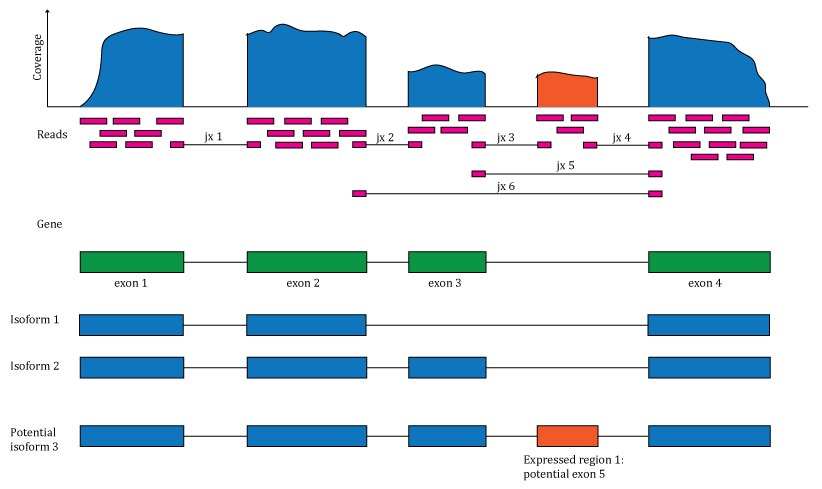
Figure 1.1: Overview of the data available in recount2 and recount3. Reads (pink boxes) aligned to the reference genome can be used to compute a base-pair coverage curve and identify exon-exon junctions (split reads). Gene and exon count matrices are generated using annotation information providing the gene (green boxes) and exon (blue boxes) coordinates together with the base-level coverage curve. The reads spanning exon-exon junctions (jx) are used to compute a third count matrix that might include unannotated junctions (jx 3 and 4). Without using annotation information, expressed regions (orange box) can be determined from the base-level coverage curve to then construct data-driven count matrices.
The following annotations are supported in recount3. See Annotation files section for direct links to the annotation files.
1.3 Study explorer
You can also open the study explorer independently through shinyapps.io.
1.4 How to cite recount3
Thank you for your continued support of the ReCount family of projects! We greatly appreciate you citing our work.
## Wilks C, Zheng SC, Chen FY, Charles R, Solomon B, Ling JP, Imada EL,
## Zhang D, Joseph L, Leek JT, Jaffe AE, Nellore A, Collado-Torres L,
## Hansen KD, Langmead B (2021). "recount3: summaries and queries for
## large-scale RNA-seq expression and splicing." _Genome Biol_.
## doi:10.1186/s13059-021-02533-6
## <https://doi.org/10.1186/s13059-021-02533-6>,
## <https://doi.org/10.1186/s13059-021-02533-6>.
##
## A BibTeX entry for LaTeX users is
##
## @Article{,
## title = {recount3: summaries and queries for large-scale RNA-seq expression and splicing},
## author = {Christopher Wilks and Shijie C. Zheng and Feng Yong Chen and Rone Charles and Brad Solomon and Jonathan P. Ling and Eddie Luidy Imada and David Zhang and Lance Joseph and Jeffrey T. Leek and Andrew E. Jaffe and Abhinav Nellore and Leonardo Collado-Torres and Kasper D. Hansen and Ben Langmead},
## year = {2021},
## journal = {Genome Biol},
## doi = {10.1186/s13059-021-02533-6},
## url = {https://doi.org/10.1186/s13059-021-02533-6},
## } |